Registries
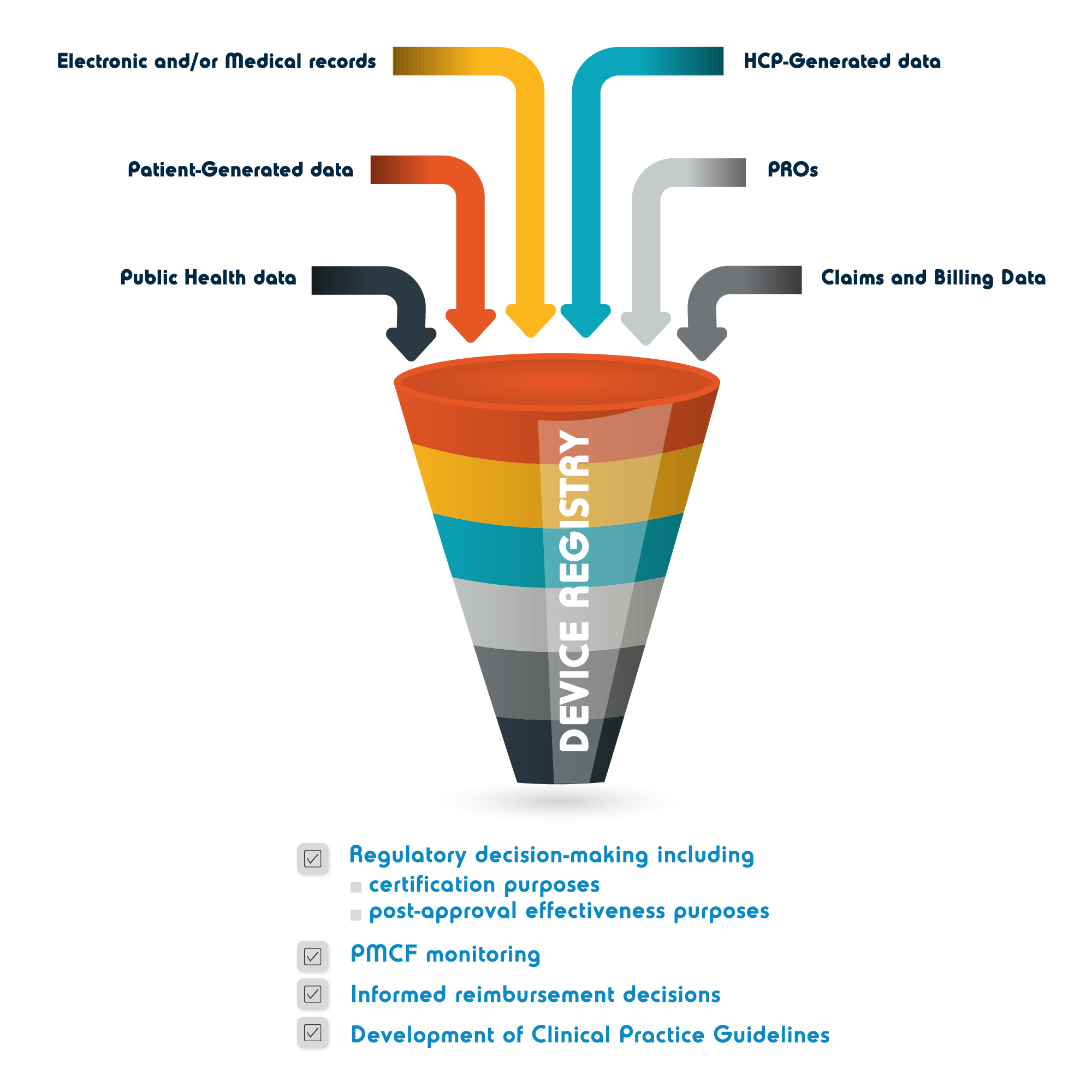
The IMDRF[1] defines a medical device registry as Organized system with a primary aim to improve the quality of patient care that continuously collects relevant data, evaluates meaningful outcomes, and comprehensively covers the population defined by exposure to particular device(s) at a reasonably generalizable scale (e.g., international, national, regional, and health system)
[1] Methodological Principles in the Use of International Medical Device Registry Data; IMDRF Patient Registries Working Group, March 2017: see it online