To Strive. To Seek. To Find.
Evnia introduces a paradigm shift in the MedTech/BioTech consulting services by offering expert medical device lifecycle management services.
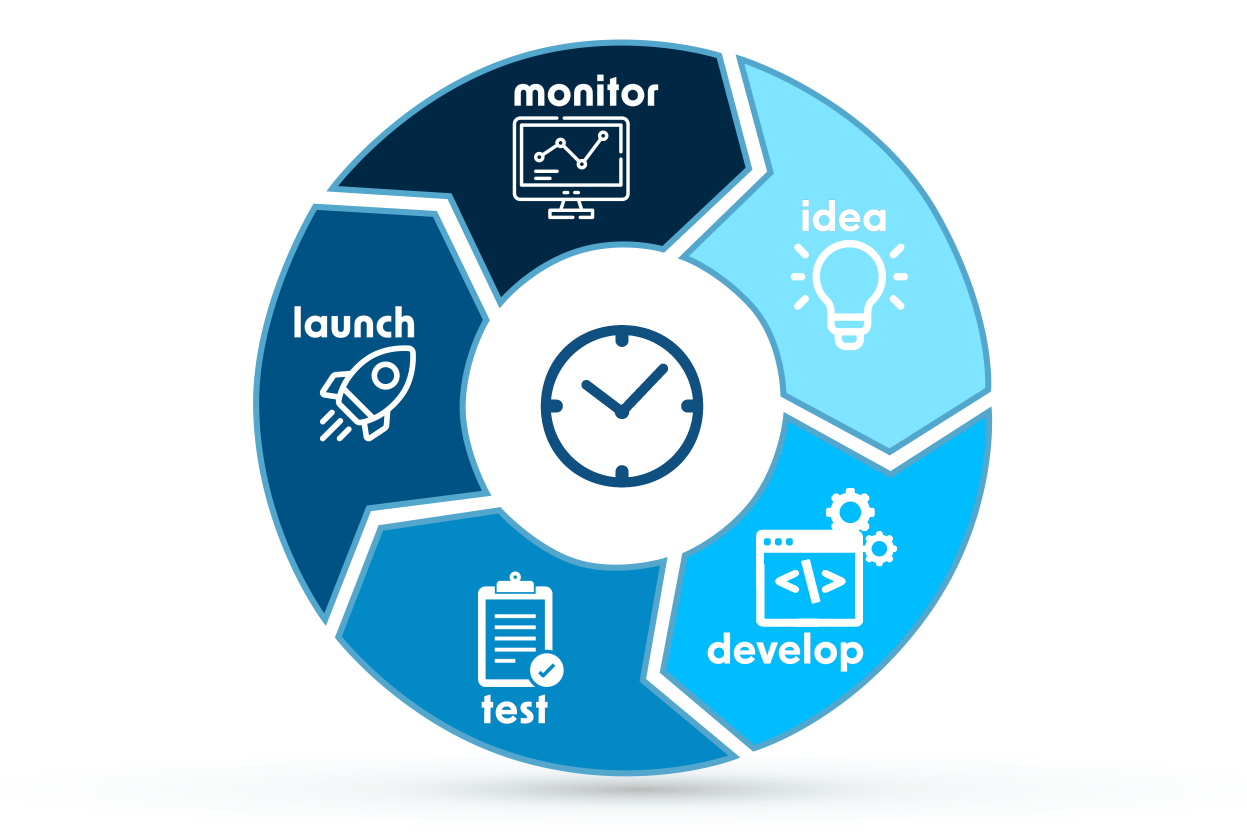
We support manufacturers throughout the medical device development roadmap. From the early concept and design stages to verification and validation, until market access and post-market adulthood, Evnia enables compliance and ensures audit and inspection readiness.
At Evnia, we serve MedTech and BioTech companies of all sizes, including Billion+, Midsize, and Startups, while remaining flexible to the needs and integration each one of our partners requires.
From building long-term partnerships to developing regulatory and clinical strategies, Evnia recognises each medical product’s unique needs and offers customised services that are adding clinical and commercial value to your portfolio. We shorten your time until market access and we support your day-to-day business pre- and post- market.
Using industry knowledge, proprietary tools, and streamlined processes, we help you turn challenges into opportunities.
Don’t know how to turn the sketch on your napkin into a successful medical product? Contact Evnia today and take your idea to the international markets faster, with less risk making patient impact.